NUR 2234: Advanced Pharmacology
Module 1
MODULE 1:
PHARMACODYNAMICS AND PHARMACOKINETICS
Learning Objectives
- Distinguish between pharmacodynamics and pharmacokinetics
- Describe drug movement throughout the body
- Identify barriers and obstacles for drugs in the body
- Explain concepts of kinetics and their clinical importance
- Identify variables affecting kinetics and their relation to drug dosing
- Explain toxicities and their relation to on-target or off-target responses
Table of Contents
- Drug-Receptor Model
- Protein Structure
- Drug Binding
- Active Site
- Drug Design
- Receptor Types
- Cellular Regulation
- Self Check Questions: Drug-Receptor Model
- Pharmacodynamics
- Dose Response Relationships
- Types of Agonists/Antagonists
- Pharmacokinetics
- Kinetic Overview
- Absorption
- Distribution
- Metabolism
- Excretion
- Half-Life (T 1/2)
- Therapeutic Dosing and Frequency
- Pharmacogenomics
- Drug-Drug Interactions
- Drug Toxicity
- Drug Toxicity Overview
- Mechanisms
- Metabolites
- Pathology of Drug Toxicity
- Carcinogenisis/Teratogenisis
- Module 1 Case Studies
Drug-Receptor Model
Protein Structure
Theoretically drugs can bind to almost any 3-D target. Desired effects (as well as adverse effects) occur when drugs bind to selected target molecules or receptors. This binding causes biochemical or physiological changes within the body. Receptors are macromolecules that mediate the biochemical or physiological changes that occur when the drug binds.
Receptors are generally proteins. Proteins have 4 major levels of structure:
- Primary
- Secondary
- Tertiary
- Quaternary
Different parts of the protein have different affinities for water:
Let's take a look at how protein structure develops:
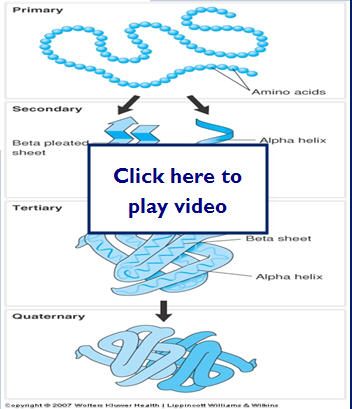
Drug Binding
The binding site on a receptor is where the drug actually binds. Binding is affected by many different types of interactions:
- van der Waals forces
- Hydrogen bonding
- Iononic interactions
- Covalent bonding (rare) is usually irreversible and creates an inactivated complex
- Drugs that act this way are sometimes referred to as suicide substrates
Now let's view an animation on drug receptor binding:
The animation above shows how drugs bind to cells.
The combination of all the interactions are what gives the drug its specificity. The favorability of the drug-receptor interaction is the affinity of the drug for the receptor (more later). Binding is rarely caused by one interaction. Stable Drug-receptor complexes usually have multiple interactions.
The molecular structure of a drug is what gives the drug its physical and chemical properties that determine how it will bind to the receptor. Important factors are:
- Hydrophobicity
- Ionization state (pKa)
- Conformation and stereochemistry
Warfarin-S enantiomer 4x more potent vs. R enantiomer due to it's ability to bind to the receptor.
Let's take a look at a drug molecule as it binds to its receptor in a protein. In this case it inhibits the enzymes action, but drugs can also stimulate action.
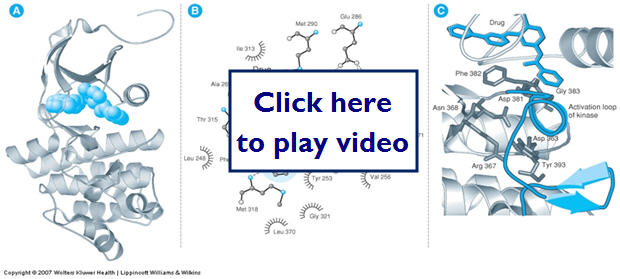
Active Site
In some cases drugs bind to sites that have enzymatic activity. This active site is where enzymatic transformation is catalyzed. If the drug inhibits binding of the normal substrate is inhibits the enzymes action. In other cases the drug may not bind to the active site.
In these cases the drug may change the shape of the receptor to increase or decrease substrate binding. Drugs can also bind and change the conformation of the receptor referred to as induced fit:
- Inactive (closed)
- Active (open)
- Desensitized (inactivated)
The structure of a drug can affect its ability to get to the receptor because of the plasma membrane. Highly water-soluble drugs are generally less able to pass through the plasma membrane or they need membrane channels or transporters. Highly lipophilic drugs cross easily and have good access to intracellular targets.
Drug Design
- Rational Drug Design
- gain knowledge of the structure of the receptor
- understand the factors affecting the receptor in order to determine the specificity between the drug and receptor
- increase efficacy and decrease toxicity by binding more selectively to the receptor
Let's now look at the drug receptor relationship in an equation type relationship:
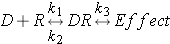
k1 = association constant (drug on)
k2 = dissociation constant (drug off)
k1 / k2 = AFFINITY
k3 = EFFICACY
This equation represents a drug binding to a receptor and forming a drug receptor complex. Please note this is not a static process and may or may not elicit a response. We will talk more about this later in pharmacodynamics.
- Determinants of drug selectivity are:
- cell-type specificity of receptor subtypes
- cell-type specificity of receptor-effector coupling.
In general, the more restricted the cell-type distribution, the more selective the drug will be, and the more receptor-effector coupling mechanisms differ among the cell types targeted, the more selective the drug will be.
Receptor Types
- Types of drug-receptors are the following:
- Transmembrane ion channel
- Transmembrane linked to intracellular G protein
- Transmembrane with enzymatic cytosolic domain
- Intracellular
- Extracellular enzyme
- Adhesion
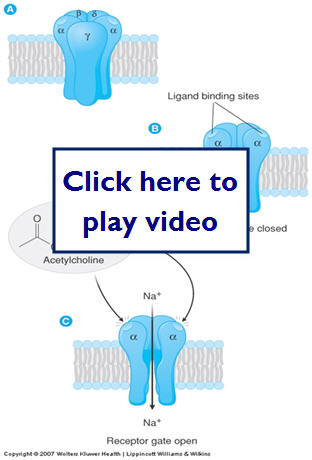
Now we can take a look at 4 common types of receptors and get a visual idea of the process of drug binding to its receptor:

- Transmembrane ion channels
- Major mechanism to regulate
- Ligand-gated
- Voltage gated
- Second messenger regulated
Let's look at regulation of an ion channel. This is an example of a Ligand-gated.
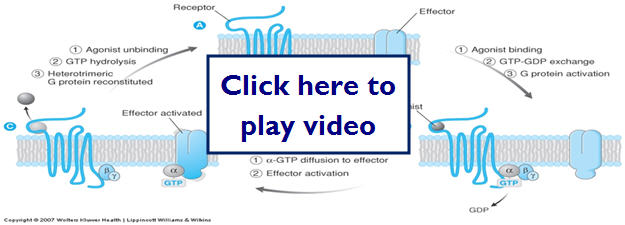
- Transmembrane G protein-coupled receptors
- Most abundant class of receptors in humans
- Exposed at the surface of the cell membrane
- Traverse through the membrane
- Have intracellular regions that activate signaling molecules called G proteins
- G proteins bind to GTP and GDP
- A major role for G proteins is to activate the production of second messengers
Now let's look at G-protein coupled receptors and what happens when an agonist binds to the receptor:
- Transmembrane receptors with enzymatic cytosolic domains
- 5 major types
- Tyrosine kinases (largest group)
- Tyrosine phosphatases
- Tyrosine kinase-associated receptors
- Receptor serine/threonine kinases
- Receptor Guanylyl cyclases
- Have roles in cell metabolism, growth and differentiation
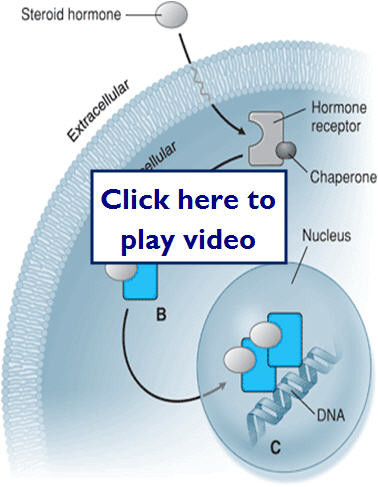
- Intracellular receptors
- Intracellular targets can be enzymes
- Alter signaling or metabolic molecules
- Lipophillic drugs can target cytosolic receptors that affect transcription regulatory factors
- Can also act on cytosolic structural proteins
- Antimicrobial and antineoplastic chemotherapy
Here we'll take a look at how a steroid hormone can diffuse through the cell membrane to act at an intercellular target receptor:
- Extracellular enzymes
- Target is outside the plasma membrane
- Can target enzymes that convert or modify molecules that can influence physiological processes
- ACEIs (inhibit angiotesin I to angiotesin II)
- Cell surface adhesion receptors
- Adhesion is when 2 cells have an area of contact between them.
- This adhesion is required for some functions to occur and is mediated by receptors
- Formation of tissue
- Migration of immune cells to a site of inflammation
Cellular Regulation
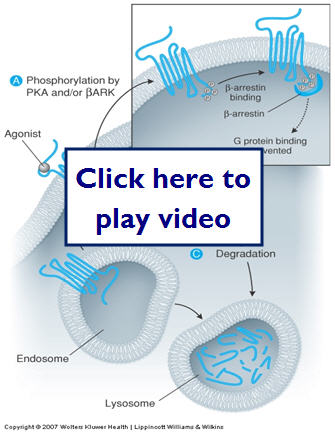
- Cellular regulation
- Overstimulation or inhibition of receptors can lead to cell damage or adverse effects to the body as a whole.
- Over time the receptors may become desensitized to the drug ( decreased ability to respond to stimuli).
- Homologous-only one type of receptor diminished
- Heterologous-two or more
- Receptors can also become inactivated
- Receptor becomes completely turned off
- Receptors may also have altered refractory times (refractoriness)
- Time before receptor can be stimulated again
- Voltage gated ion channels
- Downregulation-repeated or persistent DR interaction causes a removal of receptors of sites available for interaction
- Endocytosis sequesters the receptors
- Cells can also alter the level of synthesis of receptors, which also regulates the number of receptors available for binding
Now let's take a look at how cells can up regulate and down regulate receptors:
Not all drugs interact with receptors. Osmotic diuretics act on ion channels and change osmolarity of nephron directly. Antacids absorb or chemically neutralize acid.
Self Check Questions: Drug-Receptor Model

Pharmacodynamics
- Pharmacodynamics-the effects of a drug on the body
- Drug-receptor
- Needed to determine
- Dose ranges
- Compare potency
- Efficacy
- safety
Dose Response Relationships
- Relationship between dose (drug concentration) and the PTs response to the drug.
- In general the response to a drug is proportional to the concentration of receptors that are occupied by the drug.
Let's look at what dose-relationship curves look like on a linear and semi-logarithmic scale:
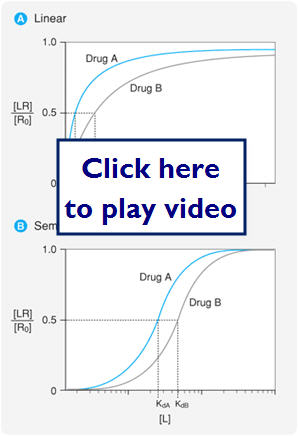
- 2 major types of dose-response relationships
- Graded-effect that various doses (concentrations) of drug have on an individual
- Quantal-effect of various doses on a population of individuals ( looks at a response to the dose)
- Sleep or no sleep, alive at 6 months or dead at 6 months
Let's first look at G-graded dose response curves:
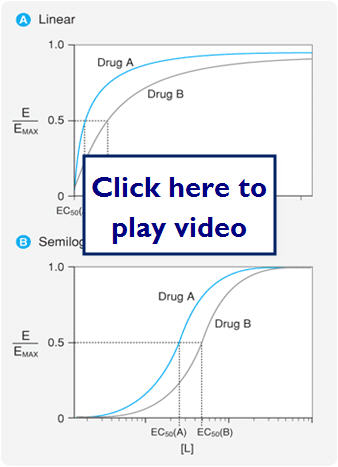
- 2 important parameters
- Potency (EC50)- concentration at which the drug elicits 50% of its max response
- Can compare the potency of 2 drugs by comparing the EC50s of the 2 drugs
- ACH EC50=12 PrCH EC50=180
- 180/12=15 therefore ACH is 15x more potent than PrCH
- Efficacy (Emax) is the maximal response produced by the drug
Now we'll look at Quantal dose response curves and critical points used in determining dosing and safety:
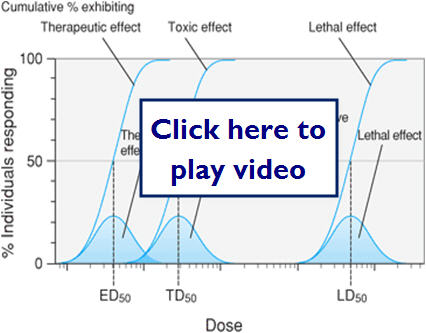
- Quantal dose-response relationships
- The responses are defined as present or not present (sleep or no sleep) versus a graded response such as heart rate.
- Important parameters for quantal responses
- Therapeutic effect
- Median effective dose (ED50)
- Adverse effect (toxic effect)
- Lethal effect
- Median lethal dose (LD50)
Let's revisit our drug receptor complex equation and the relationships it represents:
drug + receptor  drug-receptor cplx.
drug-receptor cplx.
k1 = association constant
k2 = dissociation constant
k1 / k2 = AFFINITY
k3 = EFFICACY
Types of Agonists/Antagonists
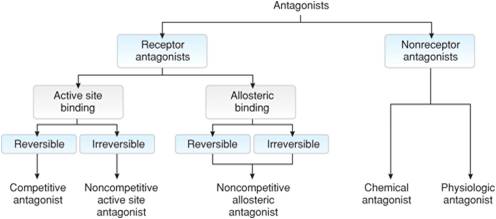
- Agonist-molecule that binds to a receptor and stabilizes it in a particular conformation (usually the active confirmation)
- Antagonist-molecule that inhibits the action of an agonist, but has no effect in the absence of the agonist
- Can bind to the receptors active site or can bind to an allosteric site
- Can be reversible or irreversible
- Competitive antagonists-binds reversible to the active site of the receptor
- Maintains receptor in the inactive conformation and blocks the agonist from binding (reduces potency), but can be pushed off receptor
- Noncompetitive antagonists-bind to either the active site or the allosteric site
- If binds to the active site effectively irreversible
- Can't be pushed off or out competed for by the agonist
- Aspirin- irreversibly inhibits cyclo-oxygenase
- If binds to the allosteric site it prevents the receptor from being activated even if the agonist binds (reduces efficacy)
- Nonreceptor antagonists
- Chemical- modifies or sequesters the agonist so it can't bind to the receptor
- Physiological- activates or blocks a receptor that mediates a physiological response opposite of the agonist
- Hyperthyroid (tachycardia)- block sympathetic stimulation
- Partial agonists-when binds to the active site only produces a partial response even if all receptors are occupied
This diagram highlights the possible site for antagonist binding:
This diagram shows different types of antagonist binding and their effect on the agonist binding:
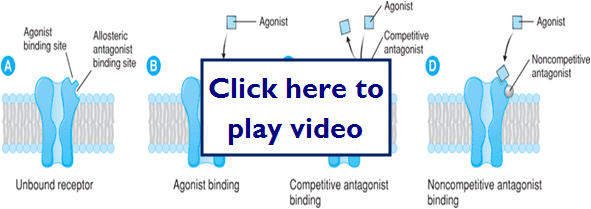
Let's take a look at a graphic representation of several types of binding and the outcomes of the overall therapeutic effect:

Let's now look at a comparative diagram of full versus partial agonists:
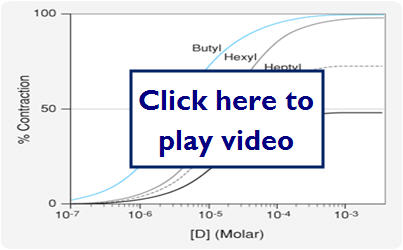
- Inverse Agonists- acts to nullify the intrinsic activity of unoccupied receptors
- When compared to competitive antagonists
- Both reduce receptor activity
- Both reduce full agonist potency
- Differ in that competitive antagonists have no effect without the presence of an agonist, whereas the inverse antagonist deactivates receptors that are normally active in the absence of an agonist
- Spare Receptors
- Situation where max effect is seen before all receptors are occupied
- Changes the dose response curve in response to a noncompetitive antagonist
- The max effect may still be achieved in response to low concentrations of an antagonist
In this diagram we will look at the effect spare receptors can have on the effectiveness of an antagonist.
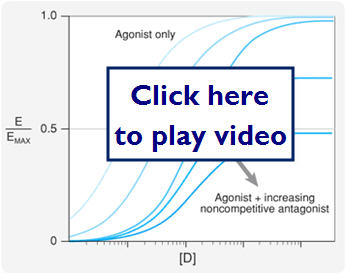
- Therapeutic window-range of drug concentrations that deliver a therapeutic response without toxicity or the drug concentration between the toxic dose and the effective dose
- Therapeutic Index-is the ratio of TD50/ED50
- Large number represents a large therapeutic index and relates the safety of the drug
- A small number indicates a narrow therapeutic index and these drugs require careful dosing and quite often monitoring for the patients safety
Self Check Questions: Pharmacodynamics
Pharmacokinetics
Kinetic Overview
- Pharmacokinetics-what the body does to the drug
- Absorption
- Distribution
- Metabolism
- Excretion
- ADME-important to keep in mind for all drugs and all PT types
We just learned that kinetics is what our body does to the drug. This diagram illustrates possible paths for a drug after it is absorbed:
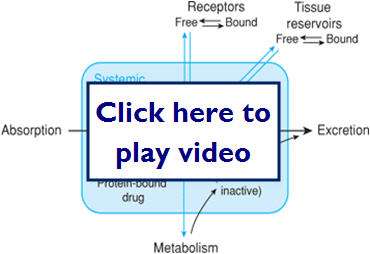
- Barriers drugs must overcome to reach the target site are
- Physical-route to enter body
- Chemical-acidic environments
- Biological-blood brain barrier, placental barrier, cell membrane
- Biological membranes
- Lipid bilayer membrane
- Hydrophillic surface with a hydrophobic lipid core
- Traversing the membranes
- The hydrophobic core of biological membranes presents the major barrier to drug transport
- Small nonpolar, lipophillic, and unionized drugs can cross via passive diffusion
- Other larger polar drugs need help
- Facilitated diffusion
- Active transport (requires energy)
- Rate of diffusion depends on the concentration gradient (Fick's Law)
- This applies when there are no complications
- Drug is small, unionized and lipophillic
- The drug will diffuse along the concentration gradient until equilibrium is reached (molecules moving in = those moving out)
PH (ION) trapping can occur when an unionized drug crosses the membrane, but due to the conditions of the environment (PH) on the other side, the molecule becomes trapped. The amount of trapped drug is determined by the pKa (dissociation constant) of the drug and the PH of the environment (NSAIDs are a good example of this).
The following illustration shows how ionization can effectively trap molecules on one side of a membrane:
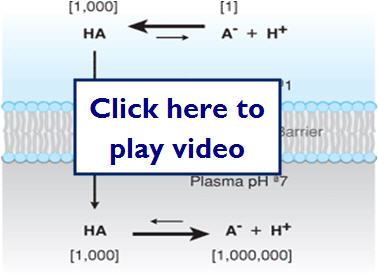
- Blood brain barrier
- Has tight junctions that does not allow most drug to pass by passive diffusion
- Drugs need to be small and lipophillic or use existing transport proteins in the blood-brain barrier to pass through
- Hydrophillic drugs can pass if they target facilitated or active transport proteins of the blood-brain barrier
- Can infuse drugs directly into the cerebrospinal fluid (CSF)
Absorption
- Absorption
- Bioavailability- amount of drug available to the target organ
- Bioavailability = quantity of drug reaching systemic circulation/quantity of drug administered (only applies to drugs that reach their target site by systemic circulation)
Let's take a look at how drugs are absorbed in the body...
Here is an example of two different drug formulations which have very different peaks:
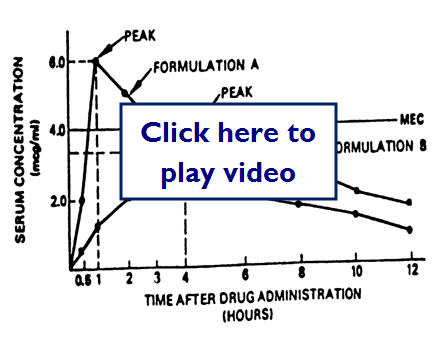
Here we have two different drug formulas but the MEC (minimum effective concentration) is lower:

Let's look at a graph comparing IV dosing, oral dosing with 100% bioavailability, and oral dosing with 50% bioavailability:
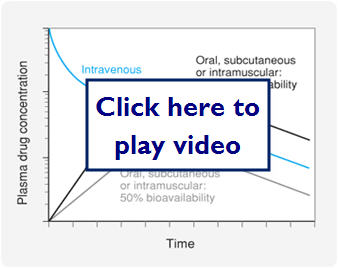
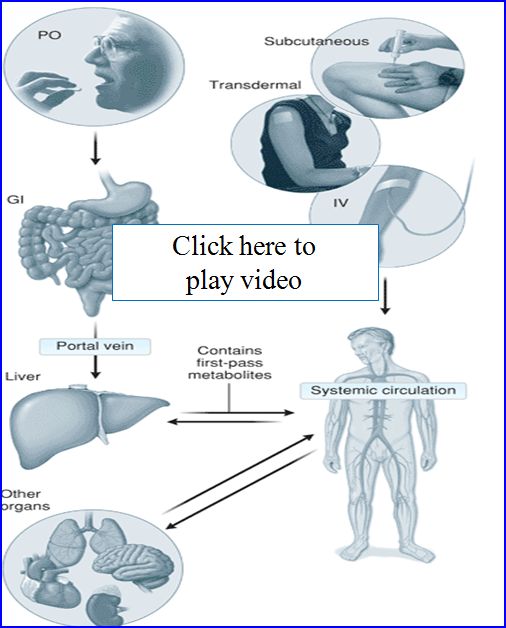
- Administration routes
- Enteral (PO)--Simplest of drug routes, but exposes drug to harsh acidic and basic environments that can limit absorption. After absorption in the GI drugs go to the liver where they experience the 1st pass effect. Liver enzymes metabolize a fraction of the drug reducing the drug concentration systemically available.
- Parenteral-Drug is delivered directly in to systemic circulation or tissue space
- SC-subcutaneous
- IM- intermuscular
- IV-intervenous
- IT-intrathecal
Let's take a look at some examples of these:
- Mucus membrane
- Sublingual, ocular, pulmonary, nasal, rectal
- Advantages
- Rapid absorption
- Convenience
- Avoid harsh GI and 1st pass effect
- Transdermal
- Absorbed through skin
- Drugs must be highly lipophillic
- Good for drugs that need slow and continuously administered
Let's look at a graph of some different bioavailabilities:

- Factors affecting absorption
- Blood flow -greatest effect
- Surface area
- High/rapid dose
Distribution
- Volume of distribution (Vd)
- Vd= Dose/(drug concentration)plasma
- Can be manipulated to find the loading dose given the Vd (can be estimated)
- Loading dose= Vd x (drug concentration) plasma SS concentration needed for therapeutic effect
- Factors that affect Vd
- Tissue distributed to (muscle, adipose, hepatic)
- Volume or mass of the tissue
- Number of binding sites in tissue
- A drug that has a higher Vd than another equally potent drug will take a higher initial dose to reach a therapeutic concentration
- Protein binding-proteins can bind (inactive)to a large percentage of a drug and only a small percentage remains free(active)
- Coadministration of other drugs can affect the percentage of bound drug (Warfarin)
Let's take a look at a video clip of volume of distribution in the body:
This diagram shows how a drug molecule crosses cell membranes and the possible routes it may take through the body to get to its target site:
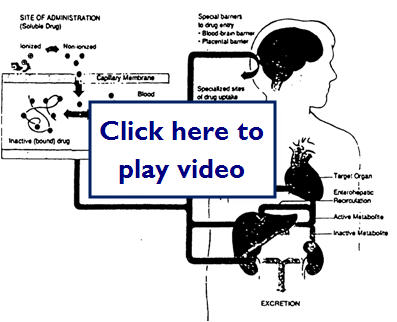
Let's take a closer look at protein binding in the following diagram:

- Kinetics of drug distribution
- Most drugs distributed rapidly from the systemic circulation to other compartments in the body
- This accounts for the sharp decrease in plasma drug concentration after an IV bolus
- The decline continues, but not nearly as fast because drug from tissues can diffuse back into blood as drug is eliminated
Let's look at a graphic representation of this:
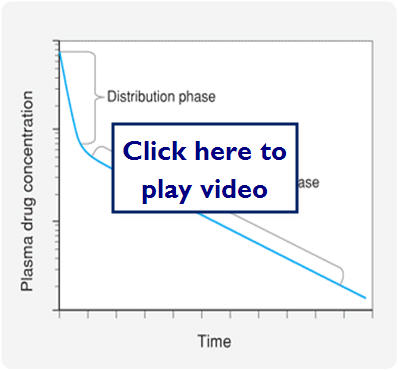
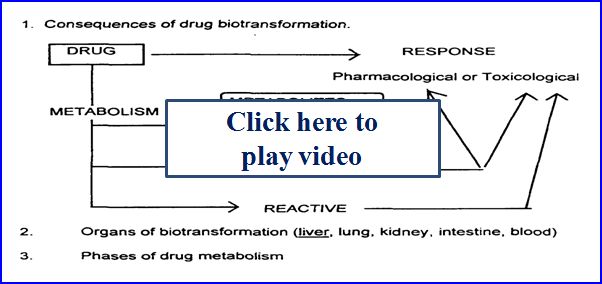
Metabolism
- Metabolism
- Liver, GI, lungs, skin, and kidneys
- Liver has the highest and most diverse metabolic enzymes
- Hepatic enzymes can chemically modify a variety of substituents on drug molecules (biotransformation), which can make the drugs inactive or facilitate its elimination
- 2 types of biotransformation
- Phase I (oxidation/reduction reactions)
- Phase II (conjugation/hydrolysis reactions)
- Oxidative/reduction reactions
- Facilitated mostly through the CYP450
- Some prodrugs take advantage of this metabolism
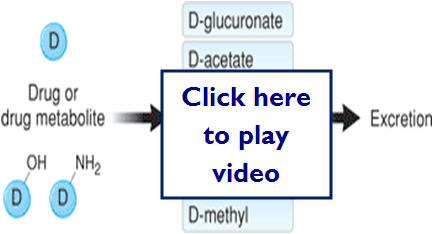
- Cojugation/Hydrolysis reactions
- Hydrolyze or conjugate a drug to a large polar molecule to inactivate or to enhance solubility and excretion in the urine or bile
- Glucuronate, sulfate, glutathione, and acetate are most commonly added
- More details next chapter
Let's take a look at how drugs are metabolized by the body:
Excretion
- Excretion
- Most drugs excreted through renal (more) or biliary (hepatic) excretion (less)
- Some unabsorbed drugs eliminated in feces
- Some respiratory and dermal
- Renal excretion
- Renal blood flow 25% of systemic blood flow
- Rate of elimination through kidneys depends on
- Filtration, secretion and reabsorption rates
Let's take a look at a diagram of where renal secretion and reabsorption take place:
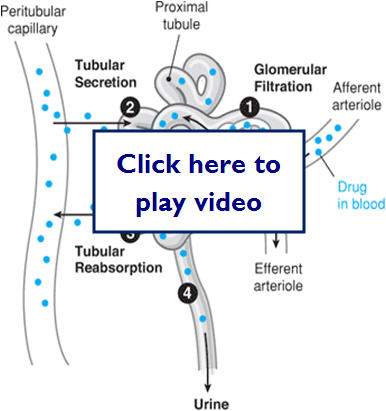
- Biliary excretion
- Drugs can be reabsorbed, because the bile duct enters the GI tract in the duodenum and drugs must pass through small and large intestine before being eliminated.
- Enterohepatic circulation-drugs get reabsorbed in the GI and retained in the portal and then systemic circulation.
Let's take a look at how drugs are excreted by the body:
Please read the following journal article to help understand why we need to be concerned with kinetics from a clinical viewpoint. Click here to download the article.
- Clinical applications
- Drug clearance is the parameter that limits the time a drug has at its molecular, cellular and organ targets. Rate of elimination of the drug from the body is relative to the concentration of drug in the plasma. Metabolism and excretion can be thought of as clearance mechanisms.
- Metabolism and Excretion kinetics
- Most drugs demonstrate first-order kinetics. Amount of drug that is eliminated in a given amount of time is directly proportional to the concentration of drug in circulation at that time (constant T1/2). Some drugs (phenytoin, ethanol) demonstrate saturation or zero order kinetics. Once saturated, the amount of drug eliminated stays constant regardless of the drug concentration in circulation. Can result in toxic or lethal effects!
Below is a comparison table of first order versus zero order drug concentrations remaining as time passes.

Half-LIfe (T 1/2)
Half-Life (T1/2) is the amount of time over which the drug concentration in the plasma decreases to ½ its original value. Because most drugs are eliminated by first order kinetics and the body can be considered a single compartment with a volume equal to that of the Vd, T1/2= 0.693xVd/clearance.
- T1/2 must be considered when designing a dosing regimen
- T1/2 of chloroquine=1 week
- T1/2 of amiodarone = 1 month
- Factors affecting T1/2
- Effects on Vd
- Aging-decreased muscle mass and distribution
- Obesity-increased adipose and distribution
- Pathological fluid-increased distribution
- Effects on clearance
- P450 induction-increased metabolism
- P450 inhibition-decreased metabolism
- Cardiac failure-decreased clearance
- Hepatic failure-decreased clearance
- Renal failure-decreased clearance
- See table 3-5 pg. 44
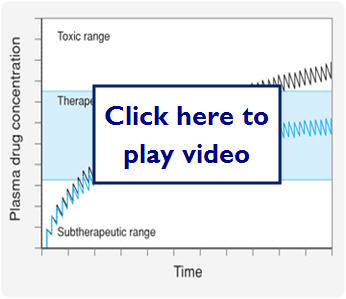
Therapeutic Dosing and Frequency
Therapeutic dosing goal is to maintain peak plasma concentration below the toxic concentration and the trough concentration above the minimum effective concentration (MEC). Steady State (SS) is reached after 4-5 half-lives for most drugs.
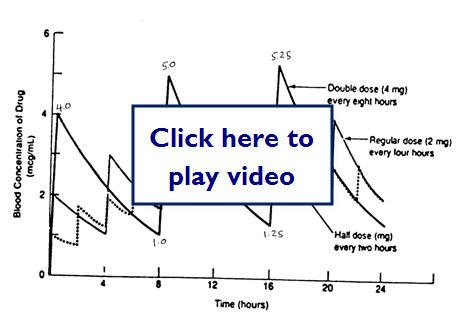
Does doubling the dose double the concentration?

Let's take a look at some examples of medication dosing:
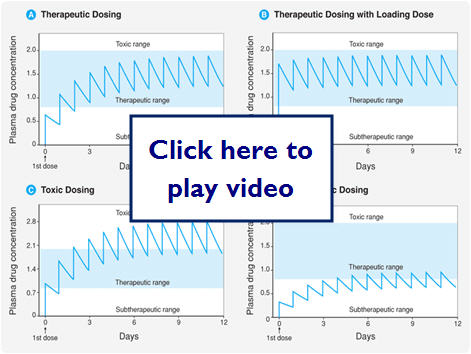
Let's look at what effect different dosing methods could have:
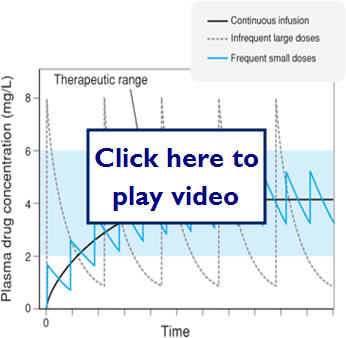
Here we see what effect zero order (saturation kinetics) can have on dosing:

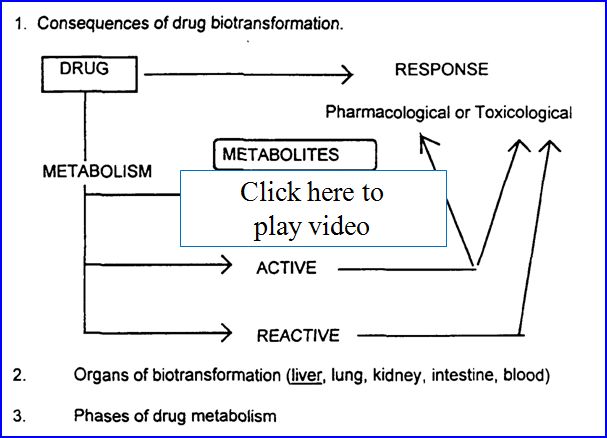
- Drug metabolism (biotransformation)
- Process by which the body alters drugs
- Drugs can be changed in 4 major ways
- Active drug to inactive drug
- Active drug to active or reactive (toxic metabolite)
- Inactive prodrug to active drug
- Unexcretable drug to excretable
Possible outcomes of metabolism:
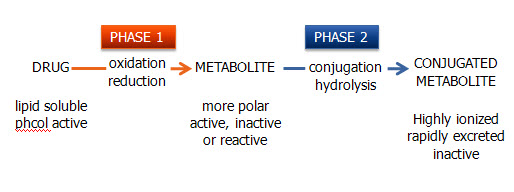
- Recall from earlier 2 major types of biotransformatiom
- Phase I (oxidation/reduction reactions)
- Phase II (conjugation/hydrolysis reactions)
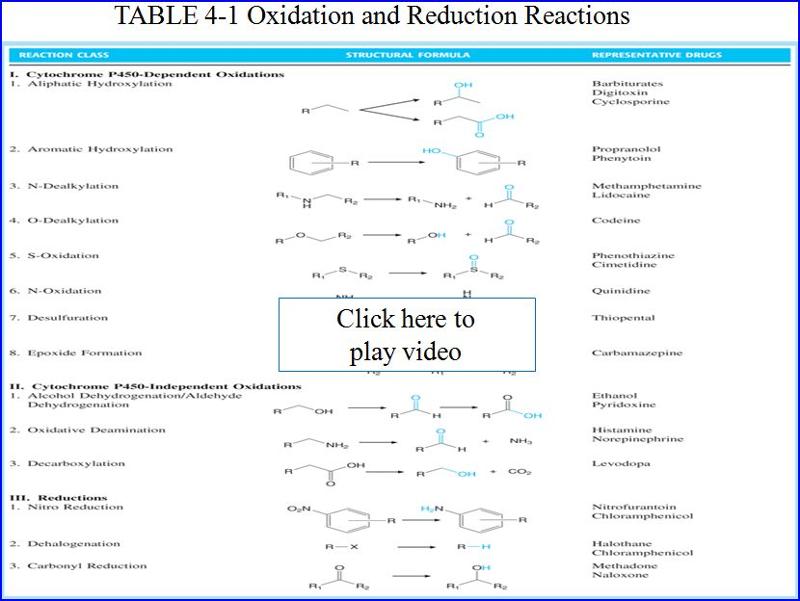
Here are some examples of Phase 1 (oxidative/reduction) metabolism by CYP 450
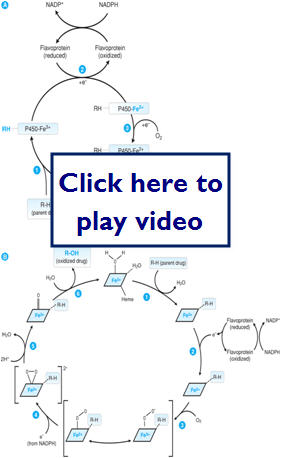
Here are some examples of Phase 2 metabolism (conjugation reaction)
- Liver is the main organ of drug metabolism
- Don't forget other organs do play roles in drug metabolism lungs, skin, GI etc.
- First pass effect
- After oral administration, drug is absorbed in the GI it gets transported through the portal vein to the liver
- As we now know the liver metabolizes drugs and therefore drugs can undergo metabolism before ever reaching the systemic circulation
- Oral Lidocaine bioavailability only 3%
Let's take a look at the first path effect and some ways to get around the problem:
- Drug transport
- Many drugs need to be actively transported into cells
- Several key molecules help facilitate this
- P-Glycoprotein
- Organic anion transporting polypeptide (OATP)
- Organic cation transporter (OCT)
- Important in 1st pass effect and statin (HMG-CoA) metabolism
- Organic anion transporter (OAT)
- Important for renal secretion (antibiotics, NSAIDs)
- Induction and Inhibition of metabolizing enzymes
- Induction- will decrease drug concentration
- Inhibition- will increase drug concentration
- Auto-induction-when drug itself induces the metabolism of itself through the enzymes
- Good tables in book on page 57,58
- Rates of biotransformation vary greatly between Pt to Pt
Here is a diagram showing enzyme induction and inhibition of cyp450 enzymes:

Pharmacogenomics
- Pharmacogenomics
- The study of genetic variability on drug metabolism
- Certain population have polymorphisms or mutations in their metabolizing enzymes, which can greatly affect drug kinetics for those PTs
- Slow metabolizers
- Fast metabolizers
- Race and Ethnicity
- Age and gender
- Diet and environment
Drug-Drug Interactions
- Drug-Drug interactions
- Increase when 2 or more drugs are administered that require metabolism via the same enzymes
- Compete for same enzyme and reduce the overall metabolism
- Can increase concentration of one or both drugs and increase adverse events
- Disease can affect metabolism
- Can change the rate or extent of drug metabolism
- Any disease of the liver can have impact on CYP450 enzymes
- Hepatitis, cirrhosis, cancer
- Cardiac disease can often decrease metabolism
- Thyroid disease can increase or decrease metabolism
Drug Toxicity
Drug Toxicity Overview
- No drug is entirely specific
- All drugs have intended and unintended effects
- unintended effects are considered side effects or adverse effects
Mechanisms
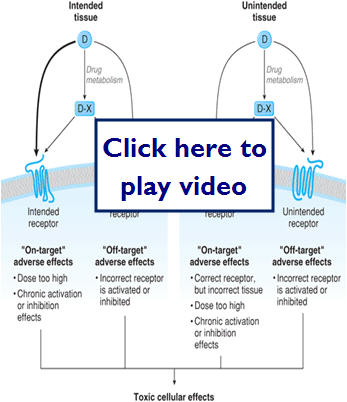
- Mechanisms of drug toxicity
- "on target"-adverse effects, drug is on right receptor, but may be wrong concentration, incorrect tissue or suboptimal kinetics
- "off target"-adverse effects, drug on wrong receptor
- Production of toxic metabolites
- Production of a harmful immune response
- Idiosyncratic responses
Let's take a look at on-target and off-target effects:
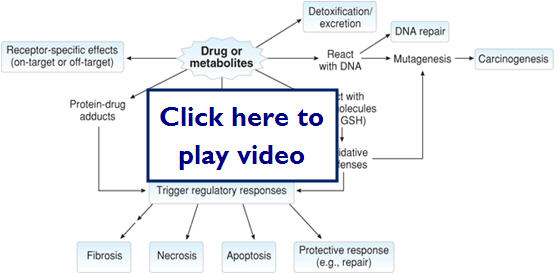
Metabolites
- Toxic metabolites
- Metabolites that are products of metabolism can be inactive, active (pharmacological effect), reactive (usually result in toxic effect)
- Harmful immune responses
- Type I
- Type II
- Type III
- Type Iv
- Drugs recognized as foreign by the immune system
- Most small molecule drug can't trigger alone
- Drug covalently bind to proteins
- Then called Haptens which can trigger immune response
Let's take a look at the 4 different types of harmful immune reactions:

- Idiosyncratic Toxicity
- No obvious mechanism is known
- Drug overdose
- Right dose is the difference between therapeutic efficacy and toxicity
- Drug-Drug interactions
- Can be due to
- enzyme metabolism
- Activation by both drug on same pathway
- Herbs can also effect
- Can be both bad and good
- Sometimes you may want to delay metabolism
- PCN and Probenecid (good)
- Sometimes drugs actively complement each other
- NTG and Viagra = severe hypotension (bad)
Pathology of Drug Toxicity
- Pathology of drug toxicity
- Can be parent drug (unmetabolized)
- Mostly metabolites that react with proteins, DNA, and oxidative defense molecules
- Can be acute or chronic
- May not be known before hand and only found out about after drug has been on the market for awhile
Here is a diagram showing the mechanisms of action of drug toxicity:
Carcinogenisis/Teratogenisis
- Carcinogenesis
- causes normal cells to transform into a neoplastic cell that expands
- Causes DNA damages or mutations
- Complex process
- Teratogenisis
- Induction of birth defects in the fetus
- Drugs that cause are called teratogens
- Please refer to Box 5-1
Module 1 Case Studies
Case 1
Mr. W. is a 66-year-old technology consultant who makes frequent trips abroad as part of his job in the telecommunications industry. His only medical problem is chronic atrial fibrillation, and his only chronic medication is warfarin. Mr. W. flies to Turkey for a consulting job. On the last night of the trip, he attends a large dinner featuring shish kebabs and other foods he does not often eat. The next day, he develops profuse, watery, foul-smelling diarrhea. A physician makes a diagnosis of traveler's diarrhea and prescribes a 7-day course of trimethoprim–sulfamethoxazole.
Mr. W. feels entirely well 2 days into the course of antibiotics, and 4 days later (while still taking his antibiotics), he entertains some clients at another lavish dinner. Mr. W. and his guests become intoxicated at the dinner, and Mr. W. stumbles and falls on the curb as he is leaving the restaurant. The next day, Mr. W. has a markedly swollen right knee that requires evaluation in a local emergency department. Physical examination and imaging studies are consistent with a moderate-sized hemarthrosis of the right knee, and laboratory studies show a markedly elevated International Normalized Ratio (INR) which is a standardized measure of prothrombin time and, in this clinical setting, a surrogate marker for plasma warfarin level. The emergency physician advises Mr. W. that his warfarin level is in the supratherapeutic (toxic) range and that this effect is likely attributable to adverse drug–drug interactions involving warfarin, antibiotics, and recent alcohol intoxication.
Case 2
You are working in a local clinic. Your patient, Mr. S, is a 65-year-old man who you have been following closely over the past few weeks for progressively elevated blood pressure readings. You have just confirmed blood pressure readings of 145/105 mm Hg in Mr. S. this morning. You decide to begin a thiazide diuretic and a beta-receptor antagonist as antihypertensive therapy. As you try to choose between atenolol and propranolol for Mr. S., you recall that the thiazide you will prescribe is 98% plasma protein bound in the blood. You also consider the following facts about the two beta-receptor antagonists.
|
|
Atenolol
|
Propranolol
|
|
Plasma protein binding
|
5%
|
95%
|
|
Urinary excretion of unchanged drug
|
85%
|
1%
|
|
Volume of distribution
|
39 L
|
270 L
|
|
β1/β2 selectivity
|
β1 selective
|
Nonselective
|